Etruscans Genetic Study
7th to 2nd centuries BC
Introduction
[(The Stanford (USA)] "publication (Achilli 2007) proves that the Etruscans are closestly related to the Turks. But given the many common words in Etruscan and Hungarian (Alinei 2003, Tóth 2007a, 2007b), the genetical relationship of Etruscan to both languages is only valid when these languages have as common basis Sumerian."
(A.Toth,
"Hungaro-Raetica",
Mikes International,
The Hague, Holland,
2007, p. 21 = >
A.Achilli et al., " Mitochondrial DNA Variation of Modern
Tuscans Supports the Near Eastern Origin of Etruscans", in
The American Journal of Human
Genetics, Volume 80 , Issue 4 , Pages 759 - 768
= > )
Like Russia a millennium later, the Romans and their descendents vehemently denied and demeaned their primogenitors for 22 centuries, and the following study must be duly credited with the balance and objectivity. The article is mirrored here because its posting on the Web can time out and disappear from the public's view.
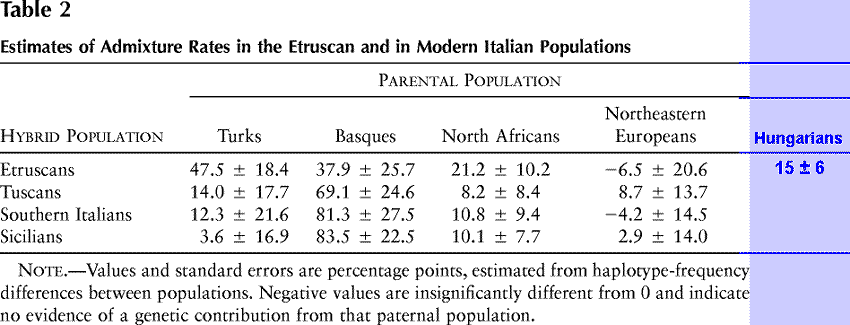
| Posting Note The original table was augmented with a blue column "Hungarians", who, based on the linguistic analyses, have at least the same rights as Sicilians and Northeastern Europeans (must be a euphemism for Finns). The difference between Finns and Hungarians is tremendous, and their common belonging to the same linguistic group may be the only common element between the two. From the very beginning of their history, Hungarians were an agglomeration of Magyar and Türkic tribes, probably a 50/50 mix; over the centuries, not too many other Magyar or Finnic people joined in, but the addition of Türkic tribes was practically nonstop: Badjanaks, Bulgars, Kumans, Kipchaks in large and small masses in the next 300 years, and many more on ad hoc basis afterwards. So, given the pre-existing Türkic population of Avars, Alans, Huns, Scythians, Sarmats, etc., the proportion of the Türkic genes must be significant, a 1/3 in the modern population would be a conservative estimate. And given that the Turkish conglomerate accumulated a measured 47.5 ± 18.4%, it would be reasonable to predict a measured 15 ± 6% for Hungarians, given all the Slavic, Germanic, and all other European genetic admixtures among the Hungarians. The number 15 ± 6% is a wild, though reasonably justified, guess. One day somebody, somehow, will check it out, and either disprove the forecast altogether, or confirm it. Linguistically the Hungarians, even though somewhat controversially, belong to the Finnic (rather, Ugro-Finnic) group, but the study is genetical, and it should have been less selective in the choice of the references. The linguistical connections between the Hungarians and Etruscans was analyzed and debated for almost a century, which makes them an undisputable candidate for the genetical comparison. A comparison with the Hungarians would significantly add to a better understanding of the composition of the Hungarians, substantiate the reasons for Etruscan-Hungarian linguistic cognates, help to understand the Türkic linguistic substrate in the Hungarian, and lead to a better understanding of the relevance of the Türkic genes in respect to Anatolian and Anatolian-unrelated component, since the Türkic admixtures to the Hungarians predate the Anatolian admixtures in genes of the Anatolian Turks. Another dimension pertains to the genetical composition of the Etruscans. The study does not aim to discern the genetic composition of the Etruscans, taking them as monolithic genetically as they were linguistically, and that implied assumption most likely is not true. Etruscans could have been as diverse as any other human society. |
This first breakthrough in the analysis of the Etruscan heritage is just a start, the authors are prudently mindful of the qualifications for the results: comparing the past with the samples of the present, absence of DNA results of the contemporaries for the same present populations, and that the admixture estimates are not to be taken at their face value because of the numerous assumptions underlying their estimation. It should be understood that the Turkish component is an ethnicity-blind property, a weighted result of the millenniums of migrations, miscegenation and extinctions of mitochondrial haplotypes, indicating a direction toward the source, but not the source itself.
Links
http://www.journals.uchicago.edu/AJHG/journal/issues/v74n4/40826/40826.html
http://www.allempires.com/forum/forum_posts.asp?TID=3521&TPN=1
Genetic Study
0002-9297/2004/7404-0009$15.00
© 2004 by The American Society of Human Genetics. All rights reserved.
The Etruscans: A Population-Genetic Study
Cristiano Vernesi,1
David Caramelli,2 Isabelle Dupanloup,1,*
Giorgio Bertorelle,1 Martina Lari,2 Enrico Cappellini,2
Jacopo Moggi-Cecchi,2 Brunetto Chiarelli,2 Loredana Castrì,3
Antonella Casoli,4 Francesco Mallegni,5 Carles Lalueza-Fox,6
and Guido Barbujani1
1Dipartimento di
Biologia, Università di Ferrara,
Ferrara, Italy; 2Dipartimento di
Biologia Animale e Genetica,
Laboratori di Antropologia, Università
di Firenze, Firenze, Italy; 3Dipartimento
di Biologia Evoluzionistica e
Sperimentale, Università di Bologna,
Bologna, Italy; 4Dipartimento di
Chimica Generale e Inorganica,
Chimica Analitica, Chimica Fisica,
Università di Parma, Parma, Italy;
5Dipartimento di Scienze Archeologiche,
Università di Pisa, Pisa,
Italy; and 6Unitat de Biologia
Evolutiva, Departament de Ciències
Experimentals i de la Salut,
Universitat Pompeu Fabra, Barcelona,
Spain
Received December 5, 2003; accepted January 28, 2004; electronically published March 10, 2004.
The origins of the Etruscans, a non-Indo-European population of pre-classical Italy, are unclear. There is broad agreement that their culture developed locally, but the Etruscans' evolutionary and migrational relationships are largely unknown. In this study, we determined mitochondrial DNA sequences in multiple clones derived from bone samples of 80 Etruscans who lived between the 7th and the 3rd centuries B.C. In the first phase of the study, we eliminated all specimens for which any of nine tests for validation of ancient DNA data raised the suspicion that either degradation or contamination by modern DNA might have occurred. On the basis of data from the remaining 30 individuals, the Etruscans appeared as genetically variable as modern populations. No significant heterogeneity emerged among archaeological sites or time periods, suggesting that different Etruscan communities shared not only a culture but also a mitochondrial gene pool. Genetic distances and sequence comparisons show closer evolutionary relationships with the eastern Mediterranean shores for the Etruscans than for modern Italian populations. All mitochondrial lineages observed among the Etruscans appear typically European or West Asian, but only a few haplotypes were found to have an exact match in a modern mitochondrial database, raising new questions about the Etruscans' fate after their assimilation into the Roman state.
* Present affiliation: Centre integratif de genomique (CIG), Lausanne, Switzerland.
Address for correspondence and reprints: Dr. Guido Barbujani, Dipartimento di Biologia, via Borsari 46, I-44100 Ferrara, Italy. E-mail: g.barbujani@unife.it
Introduction
Analysis of genetic data in modern populations has proved to be a powerful tool for reconstructing crucial aspects of human evolutionary history (Cavalli-Sforza et al. 1994; Ingman et al. 2000; Barbujani and Bertorelle 2001; Tishkoff and Verrelli 2003). It is now technically feasible to validate these analyses by directly studying the DNAs of ancient people, but so far only a handful of European sequences have been published (Handt et al. 1994; Di Benedetto et al. 2000). In this study, we present the first extensive genetic data on a European population of the preclassical period, the Etruscans.
The Etruscan culture developed in central Italy
(Etruria) in the first millennium BC
The oldest-known inscriptions in Etruscan, a
non-Indo-European language isolate, date back to the end
of the 8th century, right after the shift from the rural
Villanovian culture, documented in the same area in
the 9th century BC,
to an urban society (Bartoloni 1989).
The Etruscan cities were independent states that shared a
language and a religion but never formed a political
unit. However, between the 7th and the 5th
centuries, leagues of Etruscan cities established their political
and cultural leadership over an area spanning from
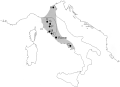
the Po Valley to Magna Graecia (fig. 1),
including, during part of the 6th century, Rome. Military
defeats, the Roman expansion, and progressive assimilation
caused a decline of the Etruscan cities, which lost their
autonomy when Roman citizenship was granted to the
Roman allies (90 89 BC).
Immediately afterward, the language disappeared (Pallottino
1975; Barker and Rasmussen
1998).
89 BC).
Immediately afterward, the language disappeared (Pallottino
1975; Barker and Rasmussen
1998).
 (28 kB) |
Figure 1 Map of Italy showing the area of Etruscan influence (gray) in the 7th and 6th centuries BC, from Barker and Rasmussen (1998). A solid line identifies the boundaries of Etruria proper. Solid circles are sampling locations: A, Adria (17 samples, 5 DNA sequences used for statistical analyses); V, Volterra (6, 3); S, Castelfranco di Sotto (2, 1); P, Castelluccio di Pienza (1, 1); M, Magliano and Marsiliana (25, 6); T, Tarquinia (18, 5); C, Capua (8, 6). Additional samples that yielded no amplifiable DNA were from Castelnuovo Berardenga (1, 0) and Pitigliano (2, 0). |
Paleoanthropological studies have only proved broad similarities between the Etruscans and their neighbors of the Iron Age (Barker and Rasmussen 1998). Archaeological evidence suggests that the Etruscan culture developed locally, with some features pointing to an Eastern influence (Pallottino 1975; Barker and Rasmussen 1998). However, it is not clear if such influence reflects only trading and cultural exchange or rather some sort of shared biological ancestry. That is a long-lasting controversy. Dionysius of Halicarnassus (1.30.2) favored local development, whereas, according to Herodotus (1.94), the Etruscans were Lydians of Anatolia who were fleeing from famine (Barker and Rasmussen 1998). No modern archaeologist supports the latter view, but some affinities between the Lydian and the Etruscan languages have been recognized (Beekes 2002), and gene flow from the eastern Mediterranean area is impossible to rule out on archaeological grounds (Tykot 1994). Unfortunately, no original documents are available to help solve this question. Indeed, although the Etruscan alphabet and language are largely understood (Bonfante and Bonfante 2002), the written record is limited to short inscriptions of religious or funerary content.
By themselves, DNA sequences cannot tell us who the Etruscans were and where they came from, but they can provide crucial information on two related questions:
2. What are the genetic relationships between the Etruscans and modern populations, and do these relationships suggest any genealogical or migrational links between the Etruscans and other Eurasians?
To address the above questions, we obtained from
museums and public collections fragments of 80
well-preserved skeletons from 10 Etruscan necropoleis (fig.
1), covering much of Etruria in terms of both
chronology (7th to 2nd centuries BC)
and geography. All human remains analyzed come from sites
where (1) the material culture has been identified
as Etruscan by archaeologists, and (2) all inscriptions,
if any, are in Etruscan. Two cities, Adria and Capua, were
at the fringes of Etruscan territory, in the Po
River valley and in Campania, respectively. Historical documents
and archaeological evidence, such as inscriptions in Etruscan
and decorations on the pottery, show that these were
indeed Etruscan settlements (Barker and Rasmussen 1998;
Bonfante 1999; Haynes
2000), although they were both
locations where hybridization may have occurred with
Veneti (in Adria) and with Samnium natives or Greek
colonizers (in Capua).
with
Veneti (in Adria) and with Samnium natives or Greek
colonizers (in Capua).
Material and Methods
DNA is generally present only in small amounts in ancient samples, and it is often damaged. Therefore, extreme precautions are needed to minimize the risk of amplifying and typing modern contaminating DNA molecules. This is especially important when dealing with relatively recent human samples, whose genetic material may not be very different from that of the archaeologists and biologists who manipulated it. To maximize the probability of extracting and sequencing authentic DNA from the ancient samples, the strictest available standards (Cooper and Poinar 2000) were followed throughout this study. All samples that did not comply with any of Cooper and Poinar's nine criteria were discarded. In most cases, no information was available on the archaeologists and museum staff who had previous contact with the bone material, but all people who manipulated the specimens for this project had their mtDNA typed and compared with the sequences obtained from the ancient specimens.
Authentication Methods for Ancient DNA
Physically isolated work area
All work was performed in isolated areas of the Florence and Barcelona laboratories, where no modern DNA has ever been introduced (criterion 1 of Cooper and Poinar [2000]).
Amino acid racemization
In living individuals, all amino acids are in the
L-enantiomeric form, but after death a racemization
process begins. Poinar et al. (1996;
Poinar and Stankiewicz 1999)
described a correlation between the amount of D-forms of
three amino acids (Asp, Glu, and Ala) and the
presence of amplifiable DNA in the sample. To test for
biochemical preservation as indirect evidence for DNA
survival, we thus estimated the degree of amino acid
racemization in each sample, using
 5
mg of bone and following the procedures described in the
work of Vernesi et al. (2001)
(criterion 7). In addition, to better estimate the
preservation of organic material, 3 mg of bone powder from
each individual was also analyzed by a thermogravimetric
assay (Peters et al. 2000).
5
mg of bone and following the procedures described in the
work of Vernesi et al. (2001)
(criterion 7). In addition, to better estimate the
preservation of organic material, 3 mg of bone powder from
each individual was also analyzed by a thermogravimetric
assay (Peters et al. 2000).
DNA extraction
After brushing and irradiating each bone surface (1 hr under ultraviolet light), DNA was extracted from powdered bone by means of a silica-based protocol (modified from Krings et al. 1997; see Di Benedetto et al. 2000). For each individual, we performed two independent extractions from different bones, usually a rib and a fragment of long bone (criterion 2). For each individual, we also attempted to amplify longer mtDNA fragments (360 bp and 700 bp), which have been reported to be unusual in ancient specimens; we sequenced only the samples in which that attempt failed (criterion 3). A negative control was included in each extraction (criterion 2).
Quantitation of ancient DNA
Sporadic contamination is considered unlikely when the number
of molecules that PCR will use as template, or
"target DNA," is > 1,000. We estimated the amount of target
DNA by competitive PCR, as described in the work
of Handt et al. (1996)
(criterion 8). A competitor was used containing a 95-bp
deletion (nucleotide positions 131225;
positions were numbered according to Anderson et al. [1981],
-16,000) in the mitochondrial hypervariable region I
(HVR-I) (Caramelli et al. 2003). PCR
components were the same as described below for the
sequencing of HVR-I, and the primers were the same as
those used for the amplification of the second of
three HVR-I fragments. A negative control was included
in each amplification (criterion 2). Thermal cycler conditions
consisted of an initial 10 min incubation at 95°C,
followed by 45 cycles of 50 s at 94°C, 50 s at
48°C, and 50 s at 72°C, with a final extension step
at 72°C for 5 min.
Amplification
The following profile was used to amplify 2
 l
of DNA extracted from the bone: 94°C for 10 min (Taq
polymerase activation), followed by 50 cycles of PCR
(denaturation 94°C for 45 s; annealing 53°C for 1 min; and
extension 72°C for 1 min) and a final step at
72°C for 10 min. The 50-l
reaction mix contained 2 U of AmpliTaq Gold
(Applied Biosystems), 200
M
of each dNTP, and 1
M
of each primer. The 360-bp-long HVR-I was subdivided in
three overlapping fragments by use of the following primer
pairs: L15995/H16132; L16107/H16261; and L16247/H16402
(Caramelli et al. 2003). Each extract was
amplified at least twice (criterion 4). Since overlapping
primers were used throughout the PCR amplifications, it is
highly unlikely that we amplified a nuclear insertion
rather than the organellar mtDNA (Handt et al.
1996; Greenwood et al.
1999).
l
of DNA extracted from the bone: 94°C for 10 min (Taq
polymerase activation), followed by 50 cycles of PCR
(denaturation 94°C for 45 s; annealing 53°C for 1 min; and
extension 72°C for 1 min) and a final step at
72°C for 10 min. The 50-l
reaction mix contained 2 U of AmpliTaq Gold
(Applied Biosystems), 200
M
of each dNTP, and 1
M
of each primer. The 360-bp-long HVR-I was subdivided in
three overlapping fragments by use of the following primer
pairs: L15995/H16132; L16107/H16261; and L16247/H16402
(Caramelli et al. 2003). Each extract was
amplified at least twice (criterion 4). Since overlapping
primers were used throughout the PCR amplifications, it is
highly unlikely that we amplified a nuclear insertion
rather than the organellar mtDNA (Handt et al.
1996; Greenwood et al.
1999).
Cloning and sequencing
PCR products were cloned (criterion 5) using the
TOPO TA Cloning kit (Invitrogen), according to the
manufacturer's instructions. Screening of white recombinant colonies
was accomplished by PCR. The colonies were transferred
into a 30-l
reaction mix (67 mM Tris HCl [pH 8.8], 2 mM
MgCl2, 1
M
of each primer, 0.125 mM of each dNTP, and 0.75
U of Taq polymerase) containing M13 forward and
reverse universal primers. After 5 min at 92°C, 30 cycles
of PCR (30 s at 90°C, 1 min at 50°C, 1 min at
72°C) were performed and clones with an insert of
the expected size were identified by agarose-gel
electrophoresis. After purification of these PCR products with
Microcon PCR devices (Amicon), a volume of 1.5
l
was cycle-sequenced, according to the BigDye Terminator
kit (Applied Biosystems) supplier's instructions. The sequence was
determined using an Applied BioSystems 377 DNA sequencer.
Independent replication
To test for contamination within the laboratory, three bone fragments were subjected to DNA extraction, amplification, cloning, and sequencing in Barcelona (criterion 6). In this lab, the following primer pairs were used: L16055/H16218 and L16209/H16401.
Associated faunal remains
The presence of human mtDNA sequences in extracts
from nonhuman bones proves that those bones have been
contaminated by human DNA and, hence, that sequences
obtained from human remains in the same burial may also
result from contamination. Cattle (Bos taurus) remains
retrieved in Magliano, in the tomb of the individual with
the 5AM sequence, gave us an opportunity to test for
this type of contamination. We tried to amplify the DNA
extracted from the cattle bones by use of both
human-specific and bovine-specific primers. We used primers
for a fragment of 152 bp of the Bos taurus
mtDNA hypervariable region and for a fragment of the human
HVR-I (the primers were L16030/H16137 and L16107/H16261,
respectively). The PCRs were performed with 2
l
of DNA, 1
M
of each primer, 200
M
of each dNTP, 1 × reaction buffer (Applied Biosystems),
1.5 mM MgCl2, and 2 U of AmpliTaq
Gold (Applied Biosystems) in a total volume of 50
l.
The cattle hypervariable region was amplified using the
following thermal cycle: initial denaturation at 94°C for
10 min, followed by 40 cycles of 94°C for 1
min, 57°C for 1 min, 72°C for 1 min, and final extension
at 72°C for 5 min. The conditions described above
for human mtDNA amplification were used in the
amplification of bovine DNA with human-specific primer.
PCR products were obtained using only the primers specific
for cattle. After run on a 1.5% agarose gel, bands of
the appropriate size were excised from the gel and
purified with Ultra Free DNA (Amicon). Cloning and
sequencing of the PCR products were performed as described
above (criterion 9).
RFLP: typing of the 14766 site
A short fragment of the coding mitochondrial region
around nucleotide position 14766 was amplified in a
50-l
reaction volume containing 2
l
of template DNA, 1.5 mM MgCl2, 200
M
of each dNTP, 2 U AmpliTaq Gold (Applied
Biosystems), and 1
M
of each primer (L14695/H14792), with the following thermal
cycles: initial denaturation at 94°C for 10 min, followed
by 40 cycles of 94°C for 1 min, 57°C for 1 min,
72°C for 1 min, and final extension at 72°C for
5 min. Fifteen
l
of PCR product were digested with 10 U of MseI
(Celbio Italy), using the recommended buffer with
overnight incubation at 37°C. Digestion products were
visualized by electrophoresis on 2.5% agarose gels. To
confirm the results of the restriction analysis, the
nucleotide at position 14766 was also determined in each
sample by independent amplification (with the same primer
pair and under the same conditions as those described
above), cloning, and sequencing.
Database of Modern Mitochondrial Sequences
The data in the database analyzed by Simoni et al. (2000) were updated with the following populations: Haviks of India (Mountain et al. 1995), Egyptians (Krings et al. 1999), Syrians and Greeks (Vernesi et al. 2001), Uighurs, Kazakhs, and Kirghiz of Central Asia (Comas et al. 1998), Armenians and Azerbaijanis (Nasidze and Stoneking 2001), and Ladin speakers of Italy (Vernesi et al. 2002), which replaced the previous sample from the same area. Populations and sample sizes are listed in the legend to figure 4.
Statistical Analysis
Haplotype variation was summarized by a reduced median network (Bandelt et al. 1999). Sites with lower mutation rates were given greater weight. Allele sharing was estimated by counting the occurrences of the haplotypes observed among the Etruscans in the 34 populations of the database.
Pairwise genetic distances (FST) between
populations were estimated by the ARLEQUIN software package
(Schneider et al. 2000),
considering both haplotype-frequency differences and numbers of
substitutions between haplotypes. Kimura's two-parameter
method was used (assuming a gamma distribution for rate
variation among sites, with
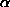 = 0.26), which allows for multiple substitutions at a site
and for different rates of transitions and transversions.
FST distances calculated in this way are
equivalent to (tm-t0)/tm,
where tm is the mean coalescence time of
two random haplotypes drawn from the two populations, and
t0 is the mean coalescence time of two
random haplotypes drawn from the same population (Slatkin
1991). From the matrix of
FST distances, we obtained a two-dimensional
representation of the data by multidimensional scaling
(MDS) (Kruskal 1964), using the
software STATISTICA.
= 0.26), which allows for multiple substitutions at a site
and for different rates of transitions and transversions.
FST distances calculated in this way are
equivalent to (tm-t0)/tm,
where tm is the mean coalescence time of
two random haplotypes drawn from the two populations, and
t0 is the mean coalescence time of two
random haplotypes drawn from the same population (Slatkin
1991). From the matrix of
FST distances, we obtained a two-dimensional
representation of the data by multidimensional scaling
(MDS) (Kruskal 1964), using the
software STATISTICA.
Patterns of genetic variation within the Etruscan and the Italian populations were described by analyses of molecular variance (AMOVA) (Excoffier et al. 1992), as implemented in the ARLEQUIN package. AMOVA estimated indexes of molecular diversity among members of the same populations, among populations, and (in the description of Etruscan diversity across time periods) among groups thereof, and tested for their statistical significance using a Monte Carlo randomization approach.
The genetic relationships between the Etruscans
and modern Italian populations were quantified by estimating
admixture coefficients for each of them, representing the
relative contribution of potential parental populations. The
purpose of this exercise was not to identify the
historical admixture process giving rise to the Etruscan
gene pool, for which the necessary data will be
available only when several ancient populations have been
described at the mitochondrial level. Rather, we wanted to
obtain figures allowing us to quantitatively compare the
composition of modern and ancient gene pools. Thus,
admixture coefficients were inferred from differences in
haplotype frequencies, considering the Etruscans and the
modern Italian populations as hybrids among up to four
potential parents (Dupanloup and Bertorelle
2001). The mitochondrial features of
the parental populations were approximated assuming that
the best available estimates of allele frequencies in past
(and unknown) populations is found in their modern
counterparts, as is customary in admixture studies (e.g.,
Chakraborty 1986; Alves-Silva et al.
2000; Chikhi et al.
2002). We chose the Basques as
representative of western Europe, the Turks as
representative of the eastern Mediterranean region, Karelians
and Volga Finns as representative of northeastern Europe,
and Egyptians and Algerians as representative of North
Africa. Admixture coefficients can also be estimated from
the coalescence times between haplotypes. However,
mitochondrial coalescence times go back to several tens of
thousand years ago in Eurasia (Richards et al.
2000). The specimens in this
study are
2,500
years old, and in this period changes of haplotype
frequencies resulting from drift certainly had a greater
effect on genetic diversity than the onset of new
mutations. Therefore, estimates based only on haplotype
frequencies seemed preferable.
Results
The 80 samples in this study underwent nine tests for authentication of ancient DNA, and at each step those that did not comply with the relevant criterion were eliminated. In particular, three samples yielded a D/L ratio for aspartic-acid enantiomers > 0.1, which is considered incompatible with retrieval of sufficient endogenous DNA from ancient remains (Poinar et al. 1996) and, hence, were discarded. In the remaining 77 samples, not only the content of racemized amino acids was low but also the relative extent of racemization was Asp > Glu > Ala, both findings generally associated with good preservation of endogenous macromolecules (Poinar et al. 1996). Low temperatures are known to facilitate the preservation of DNA in ancient samples (Kumar et al. 2000; Reed et al. 2003), and, indeed, all samples of this study come from room burials or caves, where temperatures are low and constant throughout the year.
Competitive PCR showed that 48 of these 77 samples had sufficient amounts (initial number > 1,000 copies) of template DNA (Handt et al. 1996); 29 samples were discarded at this stage. Thus, 48 samples were selected for which all tests indicated a good probability to obtain DNA sequences without artifacts. In all of them, the PCR amplification strength appeared inversely related to the size of the product, and PCR products larger than 200 bp were not observed, both findings consistent with the expected behavior of ancient (as opposed to contaminating modern) DNA molecules (Hofreiter et al. 2001). Each extract was subjected to two amplifications of three overlapping fragments, each PCR product was cloned, and multiple clones were sequenced, so that each of the three fragments was sequenced at least eight times (two extracts × two PCRs × two clones or more). Eighteen individuals yielded incomplete or multiple sequences and were discarded at this stage. Thirty individuals gave complete PCR products whose sequences were the same across clones, except for sporadic misincorporation of nucleotides in single clones. The overall misincorporation rate was a low 0.27% over the 92,104 nucleotides sequenced (compared, for example, with 0.39% in the "Iceman" specimen of Handt et al. 1994), which confirms that there was a fairly large amount of target DNA in the 30 samples.
The entire procedure was repeated in the Barcelona laboratory, under double-blind conditions, for three bone specimens, and identical sequences were obtained. Finally, the DNA from cattle remains found in the burial site of the individual with the 5AM sequence could be amplified only using bovine-specific (but not human) primers. The sequences, determined from multiple clones, match with those of the Bos taurus sequence deposited in GenBank (accession number NC_001567), showing no trace of human sequences, and so once again failing to provide evidence of modern human DNA contamination.
We observed 23 different sequences with 26
variable sites (25 transitions, 1 transversion) (table
1). (Sequences of each clone in this study can be
accessed at the authors' Web site.)
All identical sequences came from different necropoleis,
except for three individuals from the same tomb in
Tarquinia. To remove possible effects of consanguinity on
the estimated population statistics, we excluded two of
them from further analyses. A different problem was
represented by sequence 2V, which showed six substitutionsthe
069-186-189-223-319-362 motifthat
usually occur in distant and mutually exclusive branches
of the mtDNA network (Richards et al. 2000).
Independent replicates in two laboratories under
double-blind conditions confirmed the sequence, showing that
the motif does not result from experimental artifacts.
At this stage, we can only speculate that some kind
of oxydative damage, possibly resulting from the specific
microclimatic condition of the burial site (Lindahl
1993, 1997),
has altered the DNA molecules of that specimen. At
any rate, for the sake of phylogenetic consistency,
it seemed prudent not to consider this sequence in
successive statistical analyses.
 |
Table 1 Consensus HVR-I Mitochondrial Sequences in 28 Etruscan Individuals |
Exclusion of the 2V sequence brought the final sample size to 27 individuals with 22 different sequences. Only seven such sequences (4V, 5AM, 6AM, 8A, 9A, 16S, and 19M) occur in a database of 34 modern populations comprising 2,481 individuals from western Eurasia and the southern Mediterranean shores.
The Etruscan sequences show substitutions at sites (069, 126, 223, 270, and 356) known to be prone to recurrent mutation or postmortem damage (Gilbert et al. 2003). Substitutions at these mutational hotspots may cause misassignment of sequences to haplogroups (Bandelt et al. 2002). To achieve a better resolution, we then typed the restriction site of the coding region 14766 MseI, which is characteristic of the HV superhaplogroup (Richards et al. 2000). Restriction cuts on ancient DNA molecules are not always reproducible (but see Endicott et al. 2003), and so we chose not to use these markers extensively for assigning each haplotype to a haplogroup. However, the 14766 MseI restriction cuts were confirmed by sequencing of multiple clones in the region of interest. The results were unambiguous across replicates and make phylogenetic sense (with two exceptions; in these individuals, this position was left undefined) (table 1). Therefore, typing of the 14766 MseI site both clarified and further validated the results of HVR-I sequencing.
In the reduced median network based on HVR-I sequences and on the 14766 MseI polymorphism (fig. 2), several clusters are evident. Two lineages, characterized by substitutions 193-219 and 356, respectively, have a rather high internal diversity. The former substitutions are documented in Cornwall, England (but in association with 186-260-362), and the latter along the eastern and central Mediterranean shores, including Tuscany, with some derivatives in northern Europe. The only lineage containing a transversion, 095G-189, also occurs in Turkey, while the lineage with the 129 substitution is present in the eastern Mediterranean region and in northern Europe. The lineages with 126 or 126-362 presumably belong to the pre-HV haplogroup and have been observed in southeastern Europe and in the Levant.
 (46 kB) |
Figure 2 Reduced median network of the haplotypes identified among the Etruscans. Haplotype 5AM is the Cambridge reference sequence (CRS). Circle sizes are proportional to frequencies. Transitions are numbered relative to the CRS (-16,000); 095G is the only transversion. Sites undergoing recurrent mutations are underlined. The alleles shared with modern Tuscans and modern Cornish are highlighted. |
Six Etruscan sequences show the 126-193 motif. In modern individuals, these substitutions occur in lineages attributed to the J2, and occasionally T, haplogroups, accompanied by substitutions at 069 or 294, respectively, which were not observed in these six sequences. Therefore, on the basis of the HVR-I motifs, these sequences could belong to either the HV or to the JT haplogroups. However, in all of them, the MseI site was present at 14766, which rules out the hypothesis that they could belong to the HV haplogroup. Considering that postmortem changes lead to false positives (i.e., substitutions observed in the ancient sample that did not exist in the living individual) at 069 or 294 but are not known to generate false negatives there (Gilbert et al. 2003), we see no compelling reason to imagine that these results are due to postmortem damage. In addition, these sequences were determined in multiple clones for each individual, which makes a sequencing error unlikely. Therefore, despite the absence of substitutions at 069 or 294, the sequences with the 126-193 motif appear to belong to the JT haplogroup.
Internal genetic diversity within the Etruscans (gene
diversity 0.98 ± 0.01; mean number of pairwise sequence
differences 3.90 ± 2.02) is close to the average in
the database (0.97 and 4.68, respectively). AMOVA showed
no heterogeneity among Etruscan sites (1.11% of the total
[NS]), whereas differences are significant among
contemporary Italian populations (1.72% of the total, P
< .0001); in addition, we could not demonstrate any
significant differences among time periods (7th6th
centuries vs. 5th4th
vs. 3rd2nd).
None of these tests suggests that the Etruscans are more
of a cultural assemblage than a biological population.
Only two Etruscan haplotypes (5AM and 6AM, carried by 13.7% of the individuals) occur in a sample of modern Tuscans who were selected to represent inhabitants of former Etruria (Francalacci et al. 1996). The average value in comparisons of pairs of modern European populations is 27.9% ± 12.0%, showing that the genetic resemblance between the Etruscans and their modern counterparts is much less than observed between random European populations with no special evolutionary ties. Allele sharing is higher not only with the Turks (four haplotypes in common) but also with other, presumably unrelated, populations, such as the Cornish or the Germans (five and seven haplotypes in common, respectively). However, allele sharing may not be the best statistic summarizing the evolutionary relationships between the Etruscans and modern populations. Indeed, many haplotypes that so far have been observed only in the Etruscans differ by just one substitution from haplotypes that are present, or even common, among modern Europeans. Therefore, pairwise sequence differences, as well as genetic distances between populations, are more informative. The shortest genetic distances between the Etruscan and modern populations are with Tuscans (FST = 0.036; P = .0017) and Turks (FST = 0.037; P = .0001); values of FST < 0.050 were also observed for other populations of the Mediterranean shores and for the Cornish (fig. 3).
|
Figure
3 Pairwise genetic distances (FST
× 1,000) and corrected mean pairwise sequence differences
between the Etruscans and modern populations of Europe and
of the Mediterranean shores. The values referring to the
current population of Tuscany are underlined.
|
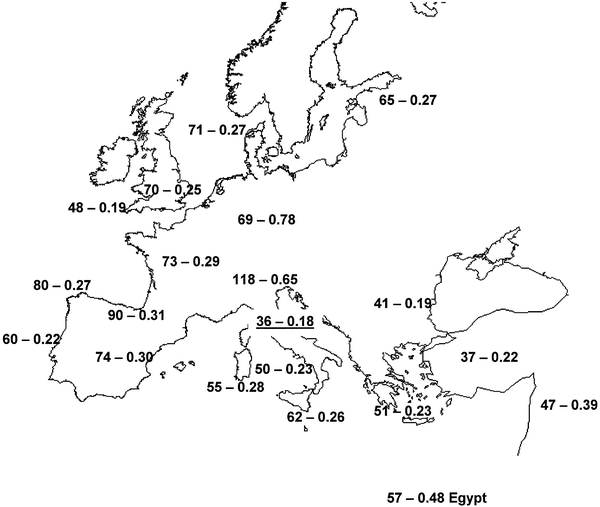
The mean pairwise sequence differences between the
Etruscans and other European populations range from 3.79
to 6.26 substitutions, with a global average of 4.64. The
Tuscans differ from the Etruscans by 4.65 substitutionsthat
is, almost exactly the value that would be expected with
a random population from the database. However, these
values are also affected by the internal diversity
of both samples being compared. If we subtract the
internal diversity, thus obtaining Nei's (1987)
dAB distance, the genetic distance
between Etruscans and Tuscans becomes the shortest, with
other southern and eastern Mediterranean populations also showing
relatively close genetic relationships (fig. 3).
In the MDS plot (fig. 4), the Etruscans
fall out of an unstructured cluster comprising most
European and Caucasus populations, including the Turks.
 (23 kB) |
Figure 4 Two-dimensional representation of the relationships between populations (MDS plot) based on FST distances. Population labels and sample sizes: ETRU: Etruscans, 27; AUST: Austrians, 117; BULG: Bulgarians, 30; DENM: Danes, 32; ESTO: Estonians, 28; FRAN: French, 111; GENO, 108: Northern Germans; GEST: Southern Germans 249; GREC: Greeks, 48; ICEL: Icelanders, 53; ITLA: Ladins of Italy, 20; ITSI: Sicilians, 63; ITSU: Southern Italians, 37; ITTU: Tuscans, 49; PORT: Portuguese, 54; SARD: Sardinians, 72; SP: Spaniards, 74; SPBA: Basques, 106; SPCA: Catalans, 15; SPGA: Gallegos, 92; SWIT: Swiss 72; UK: English, 100; UKCO: Cornish, 69; UKWA: Welsh, 92; ADYG: Adigheians, 50; ARME: Armenians, 42; AZER: Azerbaijanis, 40; CEAS: Central Asians, 100; DRUZ: Druzes, 45; EGYP: Egyptians, 91; INHA: Haviks of India, 48; NEAS: Near Easterners, 42; SYRI: Syrians, 49; and TURK: Turks, 96. |
To better compare the Etruscan gene pool with those of contemporary Italy, we treated these populations as hybrids among four potential parental populations, from the four corners of the area considered in this study (table 2). The likely contributions of each parental population, or admixture coefficients, are similar for the three modern Italian populations, but Etruscans differ in two aspects: they show closer relationships both to North Africans and to Turks than any contemporary population. In particular, the Turkish component in their gene pool appears three times as large as in the other populations. These admixture estimates are not to be taken at their face value, for numerous assumptions underlie their estimation. Here they only serve to show that, with respect to modern Italian gene pools, the Etruscan one contains an excess of haplotypes suggesting evolutionary ties with the populations of the southern and eastern Mediterranean shores.
|
Table 2 Estimates
of Admixture Rates in the Etruscan and in Modern Italian
Populations
|
Discussion
The first question we wanted to address is whether there is any evidence that the Etruscans were not a biological population, but rather an assemblage of biologically heterogeneous people who shared many cultural traits. Neither their internal genetic diversity, which is less than in comparable modern populations, nor the insignificant heterogeneity among Etruscan sites or time periods supports that view. In agreement with dental evidence (Moggi-Cecchi et al. 1997), the genetic data of this study suggest that either the people whom we call "Etruscans" shared a set of ancestors (and therefore can be considered a biological population as much as can current European populations) or they were mixed with people whose mitochondrial features did not differ from theirs.
As for the second question, which concerned the genetic relationships between the Etruscans and modern populations, various tests show that the Tuscans are the Etruscans' closest neighbors in terms of genetic distances. Despite that broad similarity, however, Etruscans and Tuscans share only two haplotypes. This finding is difficult to interpret in the absence of data on any other European population of the preclassical period. One possible interpretation is that all or most European populations of that time period were as different from their modern counterparts as the Etruscans appear to be. This would imply either extensive gene flow or a high rate of extinction of mitochondrial haplotypes, both processes causing a drastic change of the mitochondrial pool in the last 2,500 years. More importantly, a result of that kind would force us to reconsider the universally held assumption that patterns in the DNA of modern individuals reflect the evolutionary processes affecting their prehistoric ancestors (see, e.g., Piazza et al. 1988; Sokal 1991; von Haeseler et al. 1995; Richards et al. 2000, 2002; Semino et al. 2000). Alternatively, should other ancient populations prove similar to comparable modern ones, one should conclude that the Etruscans' mitochondrial sequences underwent extinction at a particularly high rate and look for an explanation for that. Until more ancient DNA data become available, both scenarios will remain possible, although we favor the latter.
Social structure may have affected these results. All skeletons we typed were found in tombs containing artifacts that could be attributed with confidence to the Etruscan culture. Those tombs typically belong the social elites (Barker and Rasmussen 1998), and so the individuals we studied may represent a specific social group, the upper classes. We do not know whether that group differed genetically from the rest of the population, which might be the case when a foreign elite imposes its rule, and often its language, over a region (Renfrew 1989). If the upper class had indeed somewhat distinct DNAs, our results could mean that this elite class became largely extinct, while the rest of the population, whose DNA we do not know, may well have contributed to the modern gene pool of Tuscany. This would be the likely effect of a process of assimilation, from which the social elites were excluded, more or less deliberately.
To summarize, only a few Etruscan sequences find an exact match in the modern database, but all belong to lineages that are still present in Europe. Some genetic affinities with modern people from western Europe reflect the sharing of lineages (5AM, 6AM, and 19M) that are widespread over the whole continent, and that therefore do not seem to point to any migrational contact but rather to a common origin of various European gene pools. On the contrary, the similarity between the Etruscan and Turkish gene pools may indeed reflect some degree of gene flow. Commercial exchanges are documented between the Etruscan harbours and Asia Minor (Tykot 1994) and trading is often accompanied by interbreeding, ultimately leading to detectable levels of genetic affinity (see Relethford and Crawford 1995). Thus, the present study suggests that gene flow from the eastern (and possibly southern) Mediterranean shores, not necessarily from Lydia as proposed by Herodotus, left a mark in the Etruscan gene pool, above and beyond what is observed in contemporary Italy.
The limited genealogical continuity between the Etruscans (be they representative of the upper class or of the entire population) and their modern counterparts of Tuscany calls for an explanation. An approximate estimate of the Etruscan population size in the 6th century BC is probably somewhere between 150,000 and 250,000 women (Rasmussen 2004). In the future, we plan to quantify the probability of extinction of all mitochondrial haplotypes but two in a subdivided population of that size, by simulating both neutral evolution and a disadvantage representing ethnic discrimination after the Roman assimilation. We shall also ask whether reasonable rates of gene flow for 2,500 years can cause a dramatic replacement of mitochondrial lineages in a population of that size. But a clearer picture is likely to emerge only when genetic data on other European populations of the same age become available for comparison.
For the time being, however, this is the first large-scale study of a pre-Roman European population in which all the strictest criteria for the validation of ancient DNA sequences have been followed. Within the limits imposed by the sample size (although our sample is large for ancient DNA studies, compared with 12 samples in Endicott et al. 2003, 19 samples in Lalueza-Fox et al. 2001, and 17 samples in Lalueza-Fox et al. 2003), the Etruscan sites appear to have rather homogeneous genetic characteristics. Their mitochondrial haplotypes are very similar, but rarely identical, to those commonly observed in contemporary Italy and suggest that the links between the Etruscans and eastern Mediterranean region were in part associated with genetic, and not only cultural, exchanges.
Acknowledgments
This study was supported by funds from the Universities of Ferrara and Florence, the Italian Ministry of Universities (MIUR [FISR and COFIN 2003]), the Fondazione Cassa di Risparmio di Ferrara, and the Fondazione Dino Terra. We are grateful to Robert Tykot, Vincent Macaulay, Peter Forster, Jaume Bertranpetit, Claudio Bravi, George van Driem, Graeme Barker, and Tom Rasmussen, for several suggestions and for critical reading of a preliminary manuscript; to S. Sanna and S. Conti, for their technical contribution; and to E. Pacciani, who provided us with the Magliano, Marsiliana, and Tarquinia samples.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- Authors' Web Site, http://web.unife.it/progetti/genetica/pdata/Etruscan.txt (for sequences of each clone in this study) First citation in article
- GenBank,
http://www.ncbi.nlm.nih.gov/Genbank/ (Homo sapiens sequences
[accession numbers AY530759AY530781]
and Bos taurus sequence [accession number NC_001567])
First citation in article
References
- Alves-Silva J, da Silva Santos M, Guimaraes PE, Ferreira AC,
Bandelt HJ, Pena SD, Prado VF (2000) The ancestry of Brazilian mtDNA lineages.
Am J Hum Genet 67:444461
First citation in article |
Full Text |
PubMed
- Anderson S, Bankier AT, Barrel BG, De Bruijn MHL, Coulson AR,
Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F Schreier PH, Smith AJ, Staden
R, Young IG (1981) Sequence and organization of the human mitochondrial genome.
Nature 290:457465
First citation in article |
PubMed
- Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks
for inferring intraspecific phylogenies. Mol Biol Evol 16:3748
First citation in article |
PubMed
- Bandelt HJ, Quintana-Murci L, Salas A, Macaulay V (2002) The
fingerprint of phantom mutations in mitochondrial DNA data. Am J Hum Genet
71:11501160
First citation in article |
Full Text |
PubMed
- Barbujani G, Bertorelle G (2001) Genetics and the population
history of Europe. Proc Natl Acad Sci USA 98:2225
First citation in article |
PubMed |
CrossRef
- Barker G, Rasmussen T (1998) The Etruscans. Blackwell, Oxford, UK First citation in article
- Bartoloni G (1989) La cultura villanoviana: all'inizio della storia etrusca. Nuova Italia Scientifica, Rome, Italy First citation in article
- Beekes R (2002) The prehistory of the Lydians, the origin of
the Etruscans, Troy and Aeneas. Biblioteca Orientalis 59:206242
First citation in article
- Bonfante G, Bonfante L (2002) The Etruscan language: an introduction. Manchester University Press, Manchester, UK First citation in article
- Bonfante L (1999) Protostoria e storia del "venetorum
angulus." Proc XX Conf Etruscan Stud, pp 501511
First citation in article
- Caramelli D, Lalueza-Fox C, Vernesi C, Lari M, Casoli A,
Mallegni F, Chiarelli B, Dupanloup I, Bertranpetit J, Barbujani G, Bertorelle G
(2003) Evidence for a genetic discontinuity between Neandertals and
24,000-year-old anatomically modern Europeans. Proc Natl Acad Sci USA 100:65936597
First citation in article |
PubMed |
CrossRef
- Cavalli-Sforza LL, Piazza A, Menozzi P (1994) The history and geography of human genes. Princeton University Press, Princeton, NJ First citation in article
- Chakraborty R (1986) Gene admixture in human populations:
models and predictions. Yearb Phys Anthropol 29:143
First citation in article
- Chikhi L, Nichols RA, Barbujani G, Beaumont MA (2002) Y
genetic data support the Neolithic demic diffusion model. Proc Natl Acad Sci USA
99:1100811013
First citation in article |
PubMed |
CrossRef
- Comas D, Calafell F, Mateu E, Perez-Lezaun A, Bosch E,
Martinez-Arias R, Clarimon J, Facchini F, Fiori G, Luiselli D, Pettener D,
Bertranpetit J (1998) Trading genes along the silk road: mtDNA sequences and the
origin of central Asian populations. Am J Hum Genet 63:18241838
First citation in article |
Full Text |
PubMed
- Cooper AR, Poinar H (2000) Ancient DNA: do it right or not at all. Science 289:1139 First citation in article | PubMed | CrossRef
- Di Benedetto G, Nasidze IS, Stenico M, Nigro L, Krings M,
Lanzinger M, Vigilant L, Stoneking M, Paabo S, Barbujani G (2000) Mitochondrial
DNA sequences in prehistoric human remains from the Alps. Eur J Hum Genet 8:669677
First citation in article |
PubMed |
CrossRef
- Dupanloup I, Bertorelle G (2001) Inferring admixture
proportions from molecular data: extension to any number of parental
populations. Mol Biol Evol 18:672675
First citation in article |
PubMed
- Endicott P, Gilbert MP, Stringer C, Lalueza-Fox C, Willerslev
E, Hansen AJ, Cooper A (2003) The genetic origins of the Andaman Islanders. Am J
Hum Genet 72:178184
First citation in article |
Full Text |
PubMed
- Excoffier L, Smouse PE, Quattro J (1992) Analysis of
molecular variance inferred from metric distances among DNA haplotypes:
application to human mitochondrial DNA restriction data. Genetics 131:479491
First citation in article |
PubMed
- Francalacci P, Bertranpetit J, Calafell F, Underhill PA
(1996) Sequence diversity of the control region of mitochondrial DNA in Tuscany
and its implications for the peopling of Europe. Am J Phys Anthropol 100:443460
First citation in article |
PubMed |
CrossRef
- Gilbert MT, Hansen AJ, Willerslev E, Rudbeck L, Barnes I,
Lynnerup N, Cooper A (2003) Characterization of genetic miscoding lesions caused
by postmortem damage. Am J Hum Genet 72:4861
First citation in article |
Full Text |
PubMed
- Greenwood AD, Capelli C, Possnert G, Pääbo S (1999) Nuclear
DNA sequences from late Pleistocene megafauna. Mol Biol Evol 16:14661473
First citation in article |
PubMed
- Handt O, Krings M, Ward RM, Pääbo S (1996) The retrieval of
ancient human DNA sequences. Am J Hum Genet 59:368376
First citation in article |
PubMed
- Handt O, Richards M, Trommsdorff M, Kilger C, Simanainen J,
Georgiev O, Bauer K, Stone A, Hedges R, Schaffner W, Utermann G, Sykes B, Pääbo
S (1994) Molecular genetic analyses of the Tyrolean Ice Man. Science 264:17751778
First citation in article |
PubMed
- Haynes S (2000) Etruscan civilization. British Museum Press, London, UK First citation in article
- Hofreiter M, Serre D, Poinar HN, Kuch M, Pääbo S (2001)
Ancient DNA. Nat Rev Genet 2:353359
First citation in article |
PubMed |
CrossRef
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000)
Mitochondrial genome variation and the origin of modern humans. Nature 408:708713
First citation in article |
PubMed |
CrossRef
- Krings M, Salem AE, Bauer K, Geisert H, Malek AK, Chaix L,
Simon C, Welsby D, Di Rienzo A, Utermann. G, Sajantila A, Pääbo S, Stoneking M
(1999) mtDNA analysis of Nile River Valley populations: a genetic corridor or a
barrier to migration? Am J Hum Genet 64:11661176
First citation in article |
Full Text |
PubMed
- Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M,
Pääbo S (1997) Neandertal DNA sequences and the origin of modern humans. Cell
90:1930
First citation in article |
PubMed
- Kruskal JB (1964) Multidimensional scaling by optimizing
goodness of fit to a nonmetric hypothesis. Psychometrika 29:127
First citation in article
- Kumar SS, Nasidze I, Walimbe SR, Stoneking M (2000)
Discouraging prospects for ancient DNA from India. Am J Phys Anthropol 113:129133
First citation in article |
PubMed |
CrossRef
- Lalueza-Fox C, Gilbert MTP, Martinez-Fuentes AJ, Calafell F,
Bertranpetit J (2003) Mitochondrial DNA from pre-Columbian Ciboneys from Cuba
and the prehistoric colonization of the Caribbean. Am J Phys Anthropol 121:97108
First citation in article |
PubMed |
CrossRef
- Lalueza-Fox C, Luna Calderon F, Calafell F, Morera B,
Bertranpetit J (2001) mtDNA from extinct Tainos and the peopling of the
Caribbean. Ann Hum Genet 65:137151
First citation in article |
PubMed |
CrossRef
- Lindahl T (1993) Instability and decay of the primary
structure of DNA. Nature 362:709715
First citation in article |
PubMed |
CrossRef
- Lindahl T (1997) Facts and artifacts of ancient DNA. Cell
90:13
First citation in article |
PubMed
- Moggi-Cecchi J, Chiarelli B, Pacciani E, D'Amore G, Brown K
(1997) The anthropological study of Etruscan populations. Etruscan Stud 4:7386
First citation in article
- Mountain JL, Hebert JM, Bhattacharyya S, Underhill PA,
Ottolenghi C, Gadgil M, Cavalli-Sforza LL (1995) Demographic history of India
and mtDNA-sequence diversity. Am J Hum Genet 56:979992
First citation in article |
PubMed
- Nasidze I, Stoneking M (2001) Mitochondrial DNA variation and
language replacements in the Caucasus. Proc R Soc Lond B Biol Sci 268:11971206
First citation in article |
PubMed |
CrossRef
- Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York First citation in article
- Pallottino M (1975) The Etruscans. Indiana University Press, Bloomington, IN First citation in article
- Peters F, Schwarz K, Epple M (2000) The structure of bone
studied with synchrotron X-ray diffraction, X-ray absorption spectroscopy and
thermal analysis. Thermochimica Acta 361:131138
First citation in article |
CrossRef
- Piazza A, Cappello N, Olivetti E, Rendine S (1988) A genetic
history of Italy. Ann Hum Genet 52:203213
First citation in article |
PubMed
- Poinar HN, Hoss M, Bada JL, Pääbo S (1996) Amino acid
racemization and the preservation of ancient DNA. Science 272:864866
First citation in article |
PubMed
- Poinar HN, Stankiewicz BA (1999) Protein preservation and DNA
retrieval from ancient tissues. Proc Natl Acad Sci USA 96:84268431
First citation in article |
PubMed |
CrossRef
- Rasmussen T (2004) Urbanisation in Etruria. In: Osborne R
(ed) Mediterranean urbanisation, 800600 BC Oxford University
Press, Oxford, UK First citation in article
- Reed FA, Kontanis EJ, Kennedy KAR, Aquadro CF (2003) Ancient
DNA prospects from Sri Lankan islands dry caves support an emerging global
pattern. Am J Phys Anthropol 121:112116
First citation in article |
PubMed |
CrossRef
- Relethford JH, Crawford MH (1995) Anthropometric variation
and the population history of Ireland. Am J Phys Anthropol 96:2538
First citation in article |
PubMed
- Renfrew C (1989) Models of change in language and
archaeology. Trans Philol Soc 87:103155
First citation in article
- Richards M, Macaulay V, Hickey E, Vega E, Sykes B, Guida V,
Rengo C, et al (2000) Tracing European founder lineages in the Near Eastern
mtDNA pool. Am J Hum Genet 67:12511276
First citation in article |
Full Text |
PubMed
- Richards M, Macaulay V, Torroni A, Bandelt HJ (2002) In
search of geographical patterns in European mitochondrial DNA. Am J Hum Genet
71:11681174
First citation in article |
Full Text |
PubMed
- Schneider S, Roessli D, Excoffier L (2000) ARLEQUIN version 2.000: a software for population genetic data analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva, Switzerland First citation in article
- Semino O, Passarino G, Oefner PJ, Lin AA, Arbuzova S, Beckman
LE, De Benedictis G, Francalacci P, Kouvatsi A, Limborska S, Marcikiae M, Mika
A, Mika B, Primorac D, Santachiara-Benerecetti AS, Cavalli-Sforza LL, Underhill
PA (2000) The genetic legacy of Paleolithic Homo sapiens in extant
Europeans: a Y chromosome perspective. Science 290:11551159
First citation in article |
PubMed |
CrossRef
- Simoni L, Calafell F, Pettener D, Bertranpetit J, Barbujani G
(2000) Geographic patterns of mtDNA diversity in Europe. Am J Hum Genet 66:262278
First citation in article |
Full Text |
PubMed
- Slatkin M (1991) Inbreeding coefficients and coalescence
times. Genet Res 58:167175
First citation in article |
PubMed
- Sokal RR (1991) The continental population structure of
Europe. Annu Rev Anthropol 20:119140
First citation in article |
CrossRef
- Tishkoff SA, Verrelli BC (2003) Patterns of human genetic
diversity: implications for human evolutionary history and disease. Annu Rev
Genomics Hum Genet 4:293340
First citation in article |
PubMed |
CrossRef
- Tykot R (1994) Sea people in Etruria? Italian contacts with
the Eastern Mediterranean in the late bronze age. Etruscan Stud 1:5983
First citation in article
- Vernesi C, Di Benedetto G, Caramelli D, Secchieri E, Katti E,
Malaspina P, Novelletto A, Terribile Wiel Marin A, Barbujani G (2001) Genetic
characterization of the body attributed to the evangelist Luke. Proc Natl Acad
Sci USA 98:1346013463
First citation in article |
PubMed |
CrossRef
- Vernesi C, Fuselli S, Castrì L, Bertorelle G, Barbujani G
(2002) Mitochondrial diversity in linguistic isolates of the Alps: a
reappraisal. Hum Biol 74:725730.
First citation in article |
PubMed
- von Haeseler A, Sajantila A, Pääbo S (1995) The genetical
archaeology of the human genome. Nat Genet 14:135140
First citation in article |
PubMed